 Search
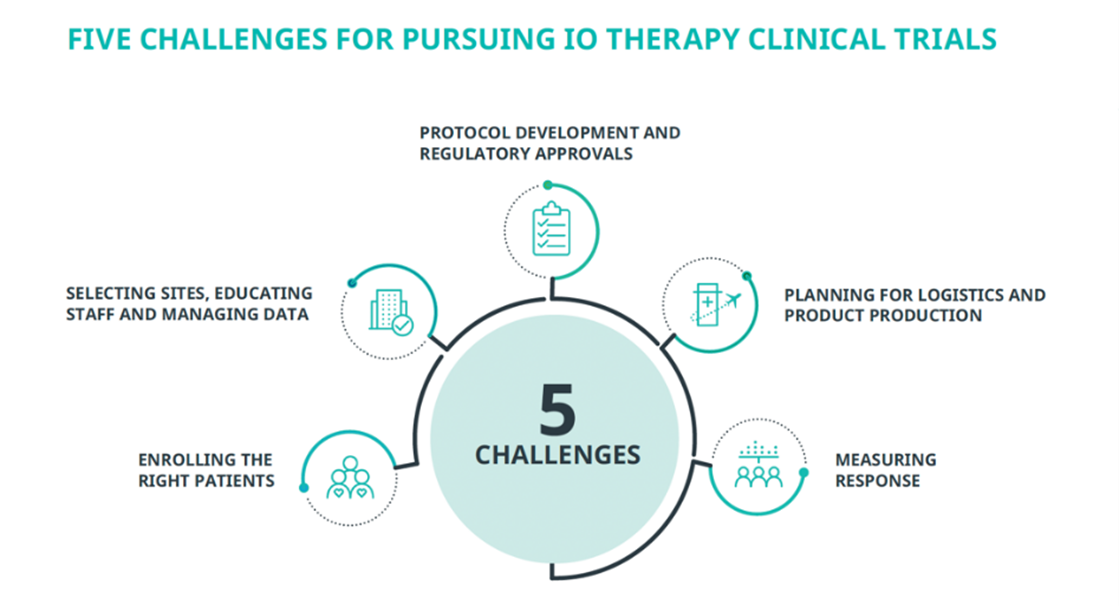
SearchImmuno-oncology (IO) therapies are transforming patient care. They also are transforming clinical research. Designing and implementing trials for cell- and gene-based therapies creates scientific and operational challenges for even seasoned sponsors. We believe the right approach to key challenges will help biotech and emerging biopharma companies successfully navigate their IO trials.
LOCAL AND CENTRAL REGULATORY APPROVALS
Being proactive with regulators helps sponsors fully understand the criteria for cell-based therapies, from CMC to pharmacology and toxicology to clinical study design and manufacturing. Via the FDA’s Project Optimus, sponsors can determine strategies to use nonclinical and clinical data during dose-finding and optimization that will later help their early registration trials limit dose toxicities that can impact efficacy evaluations and patients’ quality of life.1
At the local level, the nuanced reviews of Institutional Review Boards (IRB), Institutional Biosafety Committees (IBC) and Independent Ethics Committees (IEC) also benefit from early engagement. For example, while many multi-site trials use centralized review boards, sponsors can face additional reviews for cell therapies’ mechanisms of action, efficacy evaluations, and safety profiles. Because some IO therapies derive from recombinant nucleic acid molecules, they often require an IBC board safety approval. IBC questions can be technical, such as: “How is the vector constructed?” or “What cell lines have the vector been passaged through?” The answers typically are not in an Investigator’s Brochure, so we recommend preparing a technical document for each site-specific IBC and enlisting the preclinical or translational scientist to answer questions during reviews. We also recommend presenting an enhanced patient safety monitoring plan to the IRB/IEC.
Regulators also focus on the investigational agent’s quality and purity, and requirements vary at national or regional levels for international trials. European and China-based trials use GMP regulations, whereas the FDA and Australian Therapeutic Goods Administration recognize that GMP commercial production and warehousing regulations may not be appropriate for manufacturing phase 1 investigational agents. Due to these nuances the FDA requests product CMC information as part of IND applications.2
SELECTING SITES, EDUCATING STAFF AND MANAGING DATA
We use real world data, predictive modeling and analytics to identify and prioritize the IO-experienced sites most likely to achieve enrollment targets with diversity of clinically relevant patients. Sponsors should consider sites based on their quality of:
- processes to administer protocol-defined treatments, such as lymphodepletion chemotherapy and handling personalized therapies,
- licensing and certified capabilities to receive, handle, and ship IO agents,
- processes to manage potential adverse events and toxicities.
Even experienced sites usually require additional education to help understand the protocol’s nuances. We help sponsors create robust trial-specific instruction platforms that address patient and therapy pathways, procedures and logistics, as well as differences between the trial treatment and standards of care.
Education and platforms also help sites manage the intensive data collection and subsequent larger data casebooks of IO trials. To maintain data currency and quality, we recommend using synergistic customized solutions, such as central, remote and targeted reviews, facilitated by clearly defined data entry expectations.
ENROLLING THE RIGHT PATIENTS
IO therapy trials enroll patients defined by molecular targets such as biomarkers and genotype (their cancers’ genetic makeup) and by specific demographic criteria to match clinically relevant populations. Identifying the best molecular targets, whether proven or emerging, and finding the specific patient population requires developing a robust enrollment strategy, an ongoing monitoring plan, and careful assessment of endpoints criteria. A trial needs strategies to quantify and qualify patients’ tumor burden, side effects, immunological responses, and outcomes for supporting further development of the given IO therapy.
Because of increased sophistication in diagnostic testing during clinical care, we have found pools of pre-screened patients are more readily available. We use data and algorithms to identify high-performance sites most likely to deliver defined subsets of these pools, increasing the number of patients per site. This approach also improves study efficiency, data consistency, and speed to last-patient-in, all of which support improved IO therapies for use in clinical settings.
Notably, enrolling IO treatment-naïve patients may require fielding trials in areas where IO has not yet reached standard practice, such as selected sites in Eastern Europe, Latin America, and the Asia-Pacific region. Sponsors can benefit from CROs with in-country experience in these regions to ensure site selection for the most appropriate patients.
PLANNING FOR LOGISTICS AND PRODUCT PRODUCTION
For cell-based therapies, manufacturing brings complexities. Both autologous and allogeneic cell therapies involves supply chain considerations, including temperature control, a chain of custody, customs clearance facilitation, and regulation compliance. Moreover, the independent processing of autologous therapies requires coordinating teams across time zones and same-day handling and shipping, often involving air travel and including nights and weekends, to send patient samples to manufacturing sites and return infusions to patients. While AI can help plan and track, scheduling and coordinating require frequent real-time adjustments.
To manufacture cell-based therapies, few sponsors have in-house capabilities. We help sponsors partner with academic or commercial manufacturers with reliable track records for producing agents in the quality and quantity needed to support a trial.
Sponsors must also consider logistics and compliance of trial assessments. For example, response and safety tests use fresh blood samples with viable cells to measure cell kinetics and immune cell phenotypes. A trial using separate testing laboratories to accommodate different trial sites will require additional compliance to reduce data variability.
Our Cell and Gene Therapy Center of Excellence counsels sponsors about preclinical manufacturing and regulatory reviews. We aid preparations for the FDA’s informal consultation INitial Targeted Engagement for Regulatory Advice on CBER producTs (INTERACT) meetings and the formal IND and NDA meetings and filings.
MEASURING RESPONSE
No “universal” criteria measuring patient response to IO therapies exist in research or clinical care.3 Many sponsors augment a version of immune-related RECIST criteria with that of the Trial Reporting in Immuno-Oncology (TRIO), developed by ASCO and the Society for Immunotherapy of Cancer “to improve the interpretation and comparison of efficacy and toxicity endpoints and the combination and sequencing of treatments in IO clinical trials.”4
Typically, sponsors use objective response and progression-free survival, but overall survival is strongly suggested. Selecting the appropriate criteria for an investigational IO therapy helps determine the samples and tools that best address those criteria.
Collaborating with a knowledgeable clinical trial partner can help sponsors manage complex IO clinical trial challenges efficiently and effectively, helping them bring novel treatment options to patients with cancer.
To learn more, read our white paper: Excellence in Immuno-Oncology Clinical Trials.
1 United States Food and Drug Administration. Project Optimus. February 14, 2023. https://www.fda.gov/about-fda/ oncology-center-excellence/project-optimus. Accessed April 23, 2023.
2 United States Food and Drug Administration. Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products. March 2022. FDA-2021-D-040. https://www.fda.gov/regulatory-information/search-fda-guidance- documents/considerations-development-chimeric-antigen-receptor-car-t-cell-products. Accessed April 23, 2023.
3 Hoos A, Britten C. The immuno-oncology framework: Enabling a new era of cancer therapy. Oncoimmunology. 2012;1(3):334-339. doi:10.4161/onci.19268.
4 Tsimberidou AM, et al. Trial Reporting in Immuno-Oncology (TRIO): an American Society of Clinical Oncology Society for Immunotherapy of Cancer Statement. J Immunother Cancer. 2018; 6: 108. Published online 2018. Oct 19. doi: 10.1186/s40425-018-0426-7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6195705/ pdf/40425_2018_Article_426.pdf.